O que são craniofaringiomas?
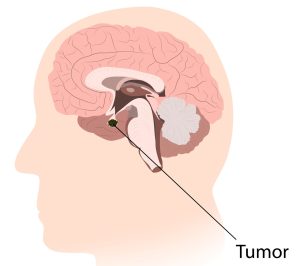
Localização do tumor craniofaringioma.
Os craniofaringiomas são considerados um tipo de tumor relativamente benigno do sistema nervoso central. As teorias relativas às causas do craniofaringioma ainda estão em estudo, mas existe uma forte corrente de médicos que acreditam que este tumor tem origem de restos embrionários de uma estrutura que existe no cérebro do bebê em desenvolvimento na barriga da mãe (a bolsa de Rathke). Por algum motivo estes restos embrionários não são eliminados como deviam e estas células futuramente poderiam dar origem a este tumor, e isso explicaria a ocorrência deste tumor na faixa etária das crianças. Para os adultos outras teorias estão em estudo, que incluiriam transformações nas células normais daquela região do cérebro que poderiam dar origem ao tumor.
Os craniofaringiomas são frequentes? Com que idade os craniofaringiomas podem aparecer?
Os craniofaringiomas são tumores pouco frequentes, com uma incidência anual de 0,5 a 2 casos por milhão de habitantes. Esses tumores são responsáveis por 1% a 3% de todos os tumores intracranianos 13% dos tumores da região supraselar (região do cérebro onde este tumor fica).
Os tumores em geral podem ocorrer em qualquer idade, mas sabemos que os craniofaringiomas tendem a aparecer preferencialmente em duas épocas da vida: a primeira entre 5 e 15 anos de idade e a segunda na 5ª década de vida (por volta dos 50-60 anos). Em crianças, o craniofaringioma representa 5-10% de todos os tumores e 56% dos tumores da região selar e supraselar. Nenhuma relação genética definitiva foi encontrada, e muito poucos casos familiares foram relatados.
Quais são os tipos de craniofaringioma? Por que é importante saber quais tipos de craniofaringioma existem?
Existem 3 tipos histológicos de craniofaringiomas: o adamantinomatoso, o papilar e o misto (uma mistura dos outros 2 tipos). O tipo adamantinomatoso é de longe o mais comum em crianças (92-96%), sendo o tipo papilar raramente relatado nesta faixa etária. A variante papilar acomete quase que exclusivamente pacientes adultos, com a média de incidência em torno de 50 anos de idade.
Em geral o tratamento das duas variantes é bastante semelhante, mas o tipo papilar tem uma taxa de recorrência muito menor do que o tipo adamantinomatoso. Isso não quer dizer que o tipo adamantinomatoso é mais agressivo, ele apenas costuma ser mais extenso e aderente, dificultando a cirurgia, de modo que há mais chance de sobrar algumas células e o tumor voltar. O tipo misto comporta-se basicamente como um tipo adamantinomatoso.
Quais os sintomas dos craniofaringiomas?
Os craniofaringiomas, assim como todos os tumores cerebrais sejam benignos ou malignos, causam sintomas por 2 motivos: compressão das estruturas cerebrais importantes que ficam próxima dele e infiltração/destruição destas estruturas.
O que temos próximo do craniofaringioma? 3 coisas importantes: a hipófise/hipotálamo (comandam quase todas as outras glândulas do organismo), os nervos ópticos (que conduzem nossa visão) e os ventrículos cerebrais, mais especificamente o 3º ventrículo cerebral.
O craniofaringioma tipicamente é um tumor de crescimento lento. Os sintomas freqüentemente se desenvolvem lentamente e geralmente se tornam óbvios somente após o tumor atingir um tamanho de cerca de 3 cm. O intervalo de tempo entre o início dos sintomas e o diagnóstico geralmente varia de 1-2 anos!
O efeito do craniofaringioma nestas estruturas pode causar um ou mais dos sintomas abaixo:
Hidrocefalia – Acúmulo de líquido no cérebro, a hidrocefalia pode estar presente tanto em crianças como em adultos.
Dor de cabeça – o sintoma mais comum, ocorre em cerca de 55-86% dos pacientes, é causada pelo crescimento do tumor apertando o cérebro ou pela própria hidrocefalia. veja mais sobre causas de dor de cabeça em crianças aqui.
Perda visual (cegueira) – também pode ocorrer em qualquer faixa de idade, em cerca de 40-70% dos pacientes. As crianças raramente se tornam conscientes de problemas visuais (apenas 20-30%) e muitas vezes a perda visual só é percebida quando já está quase completa e praticamente irreversível.
Sintomas endócrinos – Isso resulta da compressão direta ou destruição do hipotálamo e da haste hipofisária, levando à deficiência de diversos hormônios. A deficiência de hormônio do crescimento é o distúrbio mais comum endocrinológico causado pelos craniofaringiomas (35-95%) e a insuficiência adrenal secundária (deficiência de ACTH) é o segundo distúrbio mais comum (21-62% dos casos). Outros hormônios que podem ser afetados são: hormônio tireoestimulante (TSH), hormônio antidiurético (ADH ou vasopressina), hormônio luteinizante (LH) ou folículo-estimulante (FSH). Os sintomas comuns nos casos de deficiências hormonais variam de acordo com o tipo de hormônio afetado, e podem incluir: fadiga, baixa estatura (redução de crescimento na criança), diabetes insípido, ganho de peso, entre outros.
Obesidade grave – nesses casos a obesidade é acompanhada de apetite exagerado, sendo muito difícil o tratamento desses pacientes.
Disfunção sexual – 80% dos adultos queixam-se de diminuição do desejo sexual, e quase 90% dos homens se queixam de impotência, enquanto a maioria das mulheres se queixam de ausência de ciclo de menstruação.
Existe uma síndrome muito conhecida relacionada ao craniofaringioma na faixa etária pediátrica, mas embora seja bem conhecida ela é bem rara. A síndrome em questão é a Síndrome de Russel – Síndrome Diencefálica: onde as crianças apresentam perda de peso, inchaço e irritação, por vezes apresentam movimentos incomuns e involuntários dos olhos (nistagmo) e até mesmo cegueira.
Como fazer o diagnóstico de craniofaringioma?
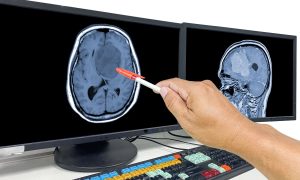
RM de crânio evidenciando craniofaringioma gigante.
A avaliação diagnóstica do craniofaringioma inclui um exame de imagem primeiramente, que pode ser a tomografia computadorizada (TC) ou, mais frequentemente, a ressonância magnética (RM). Outras avaliações são importantes, como o estudo do campo visual bem como uma avaliação endocrinológica completa.
Nos exames de imagem, existem basicamente 2 coisas que chamam a atenção nas características que sugerem os craniofaringiomas: presença de calcificações e de áreas císticas. Cerca de 80-87% dos craniofaringiomas são calcificados e 70-75% são císticos. As calcificações são mais comuns em crianças (90%) do que em adultos (50%).
Qual o tratamento para craniofaringioma? Craniofaringioma tem cura? Craniofaringioma deixa sequelas?
Essencialmente, 4 principais opções de tratamento estão disponíveis para os craniofaringiomas:
- Tentativa de ressecção total (através de cirurgia)
- Cirurgia subtotal (retirar parte do tumor), seguida de radioterapia
- Acompanhamento – tratamento conservador
- Terapêuticas intra-cisto – como bleomicina e interferon
Não existe consenso na opinião dos especialistas sobre o manejo adequado dos craniofaringiomas. A maioria dos médicos sustentam que o tratamento bem sucedido é determinado pela capacidade de preservar a capacidade cognitiva (inteligência e função social), controlar a recorrência sintomática e pela taxa de sobrevivência.
Historicamente, a proposta inicial do tratamento dos craniofaringiomas sempre foi cirúrgica, seja por via aberta (tradicional) seja pela abordagem endoscópica (endonasal). Infelizmente, a ressecção completa destes tumores é um desafio, mesmo para os neurocirurgiões experientes que operam várias crianças com craniofaringiomas a cada ano. As cirurgias radicais levam muitas das vezes ao prejuízo significativo nas funções visuais, endócrinas e cognitivas.
O objetivo do tratamento deve ser reduzir o efeito de compressão do tumor, descomprimindo vias nervosas, além de buscar recuperação da função da glândula hipofisária de maneira o mais segura possível. Complicações podem ocorrer, tais como hemorragias, cegueira e lesões na haste hipofisária com alterações hormonais permanentes, por isso o tratamento deste tumor é tão delicado.
Talvez o mais desanimador seja a ressecção total do tumor não prevenir a recorrência. Após a ressecção radical, recidivas locais são descritas em 0-60% dos pacientes. Uma vez que a cirurgia agressiva não evita recorrências carrega alta taxa de complicações, a radioterapia pós-operatória foi adicionada à cirurgia, num esforço de melhorar o controle local. A literatura médica parece apoiar esta abordagem, com um controle a longo prazo relatado de aproximadamente 80-95% em 5-20 anos e um baixo risco de complicações e sequelas definitivas. Outras terapias ainda estão em estudo para tentar melhorar essa taxa de recorrência, como a instalação de cateteres no interior de tumores císticos para aplicar medicações que ajude a controlar esta doença, como a bleomicina e o interferon.
Quando procurar um neurocirurgião pediátrico?
O neurocirurgião atua neste caso junto com diversos outros profissionais, como o endocrinologista e o neuroncologista. Nesta equipe multidisciplinar discutimos as melhores alternativas para cada paciente e individualizamos o tratamento de acordo com as características particulares de cada tumor. Tendo em vista o tratamento tão delicado deste tipo de doença, todos os esforços unidos trazem certamente um melhor resultado.
 – Agendamento de consulta de primeira vez presencial – ligação em horário comercial nos telefones – (11) 5041-1322ou (11) 5041-9988 / Agendamento automatizado pelo Whatsapp (11) 940208602.
– Agendamento de consulta de primeira vez presencial – ligação em horário comercial nos telefones – (11) 5041-1322ou (11) 5041-9988 / Agendamento automatizado pelo Whatsapp (11) 940208602.






